

Zespół rozrodczo-oddechowy świń (PRRS) nadal jest chorobą o największym znaczeniu ekonomicznym w przemyśle trzody chlewnej i jego kontrola jest dalece niewystarczająca. Lepszy i bardziej wszechstronny obraz zmienności PRRSV i monitorowanie krążenia nowych szczepów w określonym obszarze / kraju / kontynencie z pewnością pomógłby weterynarzom i hodowcom wdrożyć programy kontroli i ewentualnej eliminacji. W tym celu od końca 1990r. zaczęto stosować sekwencjonowanie PRRSV, Głównie w Ameryce Północnej, Europie i Azji Południowo-Wschodniej.
Genom PRRSV (patrz rysunek 1) jest jednoniciową cząsteczką RNA, co czyni ją podatną na "popełnianie błędów" (mutacja genetyczna) podczas replikacji w gospodarzu. Ta "tendencja do popełniania błędów" skutkuje obecnością w terenie różnych szczepów PRRSV, każdy unikalny w zakresie swojej własnej sekwencji genetycznej. To, czy różnice na tym poziomie przyczyniają się do "różnych (kliniczno-patologicznych? immunologicznych?) zachowań" nadal pozostaje przedmiotem debaty pomiędzy praktykami i badaczami.
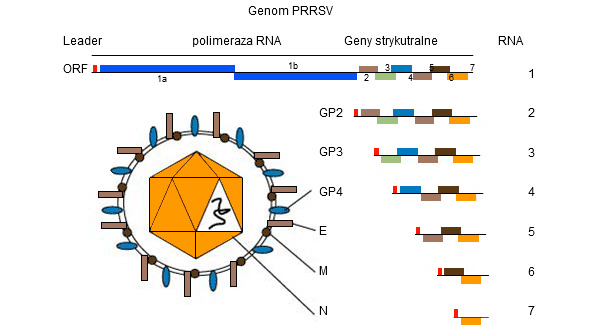
Rysunek 1. Genom PRRSV to jednoniciowa cząsteczka RNA.

Podstawy sekwencjonowania PRRSV
Sekwencjonowanie wirusa przeprowadza się na produktach PCR z próbek terenowych (surowice, tkanki, płyny ustne), uzyskując odczyt nukleotydów zazwyczaj z niektórych fragmentów genomu wirusowego RNA (patrz rysunek 2) w regionach docelowych - ORF (Open Reading Frame), a następnie porównując procentowe podobieństwo przez analizę filogenetyczną przeprowadzoną przy użyciu dedykowanego oprogramowania. Wynik tego procesu ukazuje stopień podobieństwa (homologii) między różnymi szczepami PRRSV. Za pomocą graficznego oprogramowania do wizualizacji można uzyskać również dendrogram (lub "drzewo filogenetyczne") wykazujące pokrewieństwo (lub brak) w odniesieniu do sekwencji wirusa (patrz rysunek 3).

Rysunek 2. Sekwencjonowanie wirusa przeprowadza się na produktach PCR, uzyskując odczyt nukleotydów zazwyczaj z niektórych fragmentów genomu wirusowego RNA w regionach docelowych - ORF.
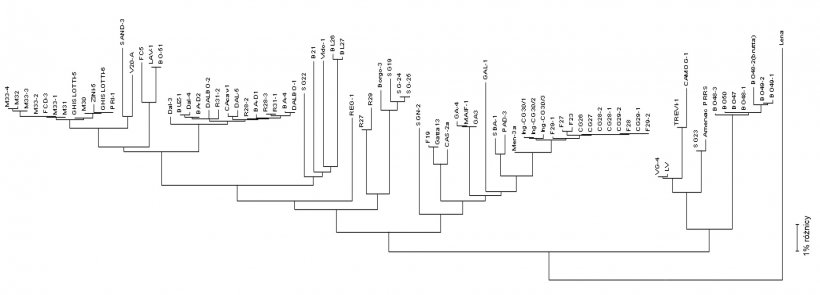
Rysunek 3. Dendrogramy lub "drzewa filogenetyczne" są używane do graficznego przedstawienia stopnia podobieństwa (homologii) między różnymi PRRSV do referencyjnej sekwencji wirusa.
Genom PRRSV koduje co najmniej 10 ORF-ów. Najczęściej stosowane do sekwencjonowania - chociaż stanowią one jedynie odpowiednio 4% i 3% całego genomu - są ORF5 (kodujące nieglikozylowane białko E) i ORF7 (kodujące białko nukleokapsydu (N)). ORF5 reprezentuje region bardziej zmienny, podczas gdy ORF7 reprezentuje region bardziej konserwatywny. Z tego powodu ten sam stopień zmienności (tj. odchylenie 5%) stwierdzone w ORF7 jest bardziej "dramatyczne" - pod względem zmiany genetycznej - w porównaniu z ORF5. Interpretacja podobieństw (tj. wirusów jest powiązanych lub nie) wymaga znacznie więcej dodatkowych informacji, ponieważ tempo zmiany genetycznej może być bardzo zmienne.
Niezwykle ważne jest zachowanie pliku rekordów wszystkich sekwencji jednoznacznie zidentyfikowanych i opatrzonych adnotacją, aby dokładnie dopasować datę, rodzaj gospodarstwa (1 lub 2 lub 3- obiektowe), przepływ trzody chlewnej, lokalizację (szerokość / długość geograficzna GPS) i pochodzenie sekwencji (rodzaj zwierzęcia / tkanka / próbka). Do tej pory nasz zestaw danych sekwencji PRRSV zawiera ponad 1300 sekwencji ORF7 od 2002 roku. Aby sensownie zinterpretować różnice, ważniejsze jest dopasowanie pojedynczych sekwencji do zdarzeń klinicznych, takich jak np. liczba ronień u loch i śmiertelność przed odsadzeniem w obiekcie 1 i śmiertelność w obiektach 2 i 3.
Pytania praktyczne
Pytania często zadawane przez hodowców i weterynarzy:
- Czy obserwowane różnice genetyczne między sekwencjami reprezentują normalną zmienność pojedynczego szczepu PRRSV w gospodarstwie / systemie produkcyjnym, czy reprezentują wiele różnych szczepów obecnych w gospodarstwie / systemie w tym samym czasie lub w krótkiej przestrzeni czasu?
- Czy mam teraz "nową epidemię" spowodowaną "nowym szczepem", czy też jest to ponowne krążenie tego samego szczepu?
Aby odpowiedzieć na te pytania, musimy zgodzić się na zaakceptowany stopień homologii dwóch szczepów wirusa zebranych w pewnym okresie czasu (12-24 miesiące?). 97-98% homologii sekwencji lub 2-3% różnicy jest wartością ogólnie przyjętą. Według mojego doświadczenia raczej trudno jest zauważyć zmianę wyższą niż 2% w "klinicznie stabilnej zamkniętej populacji" (konwencjonalne stado loch lub przepływ trzody chlewnej), ponieważ zaobserwowaliśmy uzyskanie "tego samego szczepu" przez okres 3 lat w jednym stabilnym klinicznie stadzie. Przeciwnie do tego, za każdym razem, gdy zauważyliśmy kliniczne PRRS, uzyskiwaliśmy szczep "nowy" i zmieniony filogenetycznie (90% homologii lub mniej). Niestety nie wiemy dokładnie, czy te duże różnice, które czasami obserwujemy, są wynikiem nagłej zmiany / mutacji wirusa (mało prawdopodobne w mojej osobistej opinii) lub wprowadzenia nowego szczepu. Uważa się, że podobieństwo / różnorodność genetyczna w żaden sposób nie wskazuje na podobieństwo immunologiczne (tj. wskazujące na odporność krzyżową) i nie jest w ogóle czynnikiem prognostycznym wewnętrznej patogenności (nie mówi, czy dany szczep jest "dobry" " lub zły").
Sekwencjonowanie całego genomu, które jest już w dzisiejszych czasach dostępne (niestety częściej do celów badawczych niż do codziennego użytku diagnostycznego) z pewnością pomoże odpowiedzieć na to pytanie.
Bardzo ważne jest przeanalizowanie nowych sekwencji PRRSV w oparciu o szeroki zestaw referencyjny przedstawiający gospodarstwo, system produkcyjny i region, a także sekwencje dostępnych dostępnych komercyjnie szczepionek (pozwoli to na odróżnienie szczepów terenowych od szczepionkowych). W tej chwili wciąż używamy zestawu oprogramowania open source zarządzanego przez Uniwersytet w Padova, aby budować nasze drzewa filogenetyczne. Moglibyśmy też wkrótce dołączyć do dwóch innych "programów komputerowych ad hoc" (Bioportal University of Davis-California i CLASSIFARM-PATH by IZSLER (Brescia, Włochy), które będą miały znacznie większy zestaw sekwencji do porównania z umożliwieniem lepszego zrozumienia krążenia PRRSV we Włoszech i być może w UE.
Podziękowania dla Prof. Michele Drigo (UNI-PD) za interesującą dyskusję i recenzję tego artykułu.





