

Wirus zespołu rozrodczo-oddechowego świń (PRRSV) wraz z wirusem afrykańskiego pomoru świń jest jednym z najważniejszych ekonomicznie patogenów wpływających na przemysł trzody chlewnej na całym świecie. Niedawne pojawienie się agresywnych szczepów PRRSV, takich jak wysoce patogenny PRRSV (HP-PRRSV) w Azji, Rosalia w Europie i L1C.5 w Ameryce Północnej, wywołało dyskusję na temat potrzeby dalszej poprawy diagnostyki i kontroli PRRSV.
Wykorzystanie RT-PCR do wykrywania materiału genetycznego PRRSV jest powszechnie stosowane na całym świecie do badań przesiewowych populacji pod kątem obecności wirusa.

Krokiem wykraczającym poza wykrywanie RNA jest sekwencjonowanie genomu PRRSV, powszechnie wykonywane przy użyciu techniki Sangera. Na całym świecie część wirusa najczęściej wykorzystywana do sekwencjonowania PRRSV to ORF5, chociaż niektóre laboratoria oferują również sekwencjonowanie ORF7.
ORF5 stanowi około 4%, a ORF7 2,4% genomu PRRSV, a zatem nie zapewniają one pełnego pokrycia genetycznego całego genomu.
W ostatnich latach wzrosło zainteresowanie wykorzystaniem sekwencjonowania nowej generacji (NGS) do odzyskiwania całych genomów PRRSV do badań epidemiologicznych PRRSV w stadzie lub systemie produkcyjnym (rysunek 1).
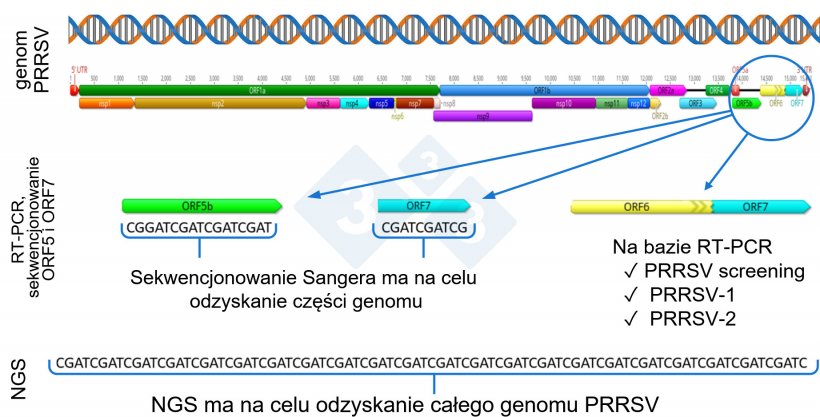
Zwykle podczas procesu replikacji PRRSV przechodzi zmiany genetyczne i mutacje, które mogą potencjalnie prowadzić do pojawienia się nowych wariantów wirusa. PRRSV jest jednym z wirusów o wysokim wskaźniku mutacji, około 0,5 do 1% rocznie, nierównomiernie rozłożonym w różnych regionach genomu, typie wirusa i liniach genetycznych, co prowadzi do ciągłej ewolucji genetycznej. Warto zauważyć, że mutacje genetyczne i ewolucja wirusa mogą zachodzić we wszystkich genach, a sekwencjonowanie tylko części genomu, np. ORF5 lub ORF7, prawdopodobnie pomija możliwość wykrycia zmian, które zaszły poza sekwencjonowanym regionem (rysunek 1).
W tej sytuacji NGS staje się użytecznym narzędziem, zapewniając możliwość odzyskania całego genomu PRRSV do wykorzystania w badaniach epidemiologicznych.
W jaki sposób lekarze weterynarii i hodowcy mogą wykorzystać NGS?
Aby zmaksymalizować użyteczność NGS, należy wziąć pod uwagę kilka kwestii:
-
Wczesne zaangażowanie lekarza weterynarii i diagnosty laboratoryjnego jest ważne, aby dostosować podejście diagnostyczne, oczekiwania i testy.
-
Czy istnieje sekwencja całego genomu szczepu referencyjnego PRRSV w gospodarstwie lub systemie produkcyjnym do porównania?
- Jeśli nie, należy użyć NGS na dodatnich próbkach PRRSV RT-PCR o niskich wartościach Ct, tj. idealnie < 24, aby odzyskać cały genom jako szczep referencyjny stada. Wartość Ct jest odwrotnie proporcjonalna do ilości kopii genetycznych wirusa obecnych w próbce, tj. im niższa wartość Ct, tym wyższa oczekiwana wiremia i potencjalny sukces NGS;
- Szczep referencyjny umożliwia późniejsze porównanie prospektywnie odzyskanych genomów w celu zrozumienia ewolucji genetycznej PRRSV w gospodarstwie, przepływie i systemie.
- Jaki jest cel wykonania NGS?
-
Jeśli celem jest wykrycie ewolucji wirusa na poziomie całego genomu: należy pobierać w płuca i surowicę, ponieważ z tych próbek z większym prawdopodobieństwem pozyska się cały genom.
-
Jeśli celem jest zrozumienie różnorodności wirusów w gospodarstwie lub stadzie, należy użyć typów próbek populacyjnych, takich jak płyn pokastracyjny lub po obcinaniu ogonków, próbki płynu ustnego lub spulowane próbki indywidualne, np. zbiorcza surowica jest bardziej podatna na odzyskanie wielu wirusów, jeśli są one obecne w próbce.
-
- Infekcje mieszane z dwoma lub więcej wirusami PRRSV w stadzie są rzeczywistością (rysunek 2a), a jeśli dwa wirusy są bardzo podobne, NGS może nie pozyskać całego genomu;
- Jeśli w próbce znajduje się wiele wirusów, NGS może odzyskać fragmenty genomu, zwane „kontigami”, z różnych wirusów, które po porównaniu ze szczepem referencyjnym gospodarstwa mogą pomóc w rozpoznaniu, czy w próbce znajduje się wiele wirusów (rysunek 2b);
- Na koniec, należy upewnić się, że jest ktoś, kto pomoże w analizie genetycznej i porównaniach oraz skoordynuje czas realizacji, ponieważ NGS jest zazwyczaj powolnym procesem, którego ukończenie może zająć ponad tydzień.
Jakiego rodzaju informacje epidemiologiczne możemy uzyskać dzięki NGS?
Wygenerowane dane wyjściowe NGS mogą pomóc w rozstrzygnięciu wątpliwości:
-
Czy wirus ewoluował poprzez losowe podstawienia nukleotydów w tych samych pozycjach genomu? Jak bardzo wirus zmienił się między dwoma punktami czasowymi i w którym genomie, tj. w regionach ORF, nastąpiły te zmiany?
-
Czy wirus ewoluował poprzez insercje lub delecje w swoim genomie?
-
Czy w gospodarstwie, stadzie lub przy okazji przemieszczania zwierząt wystąpiło nowe wprowadzenie niespokrewnionego wirusa?
-
Chociaż jest to mniej prawdopodobne, ale możliwe, czy nowy wirus nabył zmiany w regionach genomu, które są fragmentami używanymi przez sondy lub startery podczas RT-PCR lub sekwencjonowaniu Sangera, powodując fałszywie ujemne wyniki?
-
Czy wirus uległ rekombinacji, tj. nabył pewne regiony genomowe od dwóch lub więcej wirusów rodzicielskich?
Rekombinacja jest naturalnym procesem ewolucji PRRSV i występuje, gdy dwa różne szczepy PRRSV replikują się w tej samej komórce, generując trzeci szczep - wirusa pochodnego. Rekombinacja będzie zgłębiana w kolejnym artykule "Konsekwencje dylematu związanego z rekombinacją wirusa PRRSV".

