Wstęp
Wirus zespołu rozrodczo-oddechowego świń (PRRSV) posiada genom w postaci pojedynczej nici RNA co oznacza, że bardzo łatwo ulega on mutacjom. Konsekwencją tego jest fakt, że każdy szczep czy izolat PRRSV jest unikalny. Dlatego też genotypowanie szczepów PRRSV jest ważną metodą diagnostyczną w rozpoznawaniu i zwalczaniu choroby. Genotypowanie polega na oznaczaniu sekwencji nukleotydów w DNA będącym kopią fragmentu RNA genomu wirusa. Proces ten nazywamy sekwencjonowaniem DNA. Obecnie najczęściej wykorzystywanym do genotypowania fragmentem genomu PRRSV jest gen ORF5, kodujący glikoproteinę otoczki wirusa, z względu na bardzo wysoką zmienność genetyczną.

Diagnostyczne sekwencjonowanie DNA
Szczepy PRRSV Typu 1 (europejskie) i Typu 2 (amerykańskie) mogą być bardzo łatwo odróżnione od siebie na podstawie przeprowadzenia badania jednym z wielu testów PCR. Odróżnienie od siebie szczepów PRRSV należących do każdego z dwóch genotypów wymaga już sekwencjonowania DNA. W tym celu z płynów ciała lub wycinków narządów wewnętrznych zawierających odpowiednio dużą ilość wirusa, wyodrębnia się RNA, które następnie jest przepisywane na DNA (kopiowane) w reakcji odwrotnej transkrypcji, DNA to jest poddawane reakcji PCR w celu namnożenia dużej liczby kopii genu ORF5, a produkt takiej reakcji jest wysyłany do laboratorium wykonującego sekwencjonowanie. Proces ten jest w większości zautomatyzowany i na ogół zajmuje do 3 dni. Surowe dane sekwencyjne trafiają do laboratorium diagnostycznego w celu właściwej analizy. Raport z takiej analizy na ogół zawiera sekwencję nukleotydową ORF5 badanego szczepu, tabelę podobieństwa sekwencji badanego szczepu do sekwencji szczepów szczepionkowych, a niektóre laboratoria podają również podobieństwo do panelu sekwencji szczepów referencyjnych, sekwencji z danego obszaru czy systemu produkcyjnego. Wyniki takie posiadają często formę dendrogramu, czyli drzewa filogenetycznego sekwencji.
Analiza sekwencji PRRSV
Podobieństwo sekwencji jest określane w wyniku porównania dwóch lub więcej sekwencji przy pomocy programów komputerowych. Przykład porównania kilku sekwencji ORF5 (fragment) uzyskanych w pięciu różnych fermach pokazano na rycinie 1. Ich wzajemne podobieństwo wyrażone w procentach pokazano w tabeli. Sekwencje nukleotydów użyte w przykładzie są do siebie podobne w zakresie od 81,2 do 99,8%. Dendrogram skonstruowany w oparciu o te sekwencje pokazuje grupowanie najbardziej podobnych do siebie szczepów (Rycina 2).

Rycina 1: Porównanie sekwencji fragmentu ORF5 szczepów PRRSV z 5 różnych ferm. Z ferm 1, 4 i 5 uzyskano po kilka sekwencji. Kropki oznaczają pozycje w sekwencjach identyczne z pozycjami w szczepie referencyjnym PRRSV Typ 1 Lelystad.
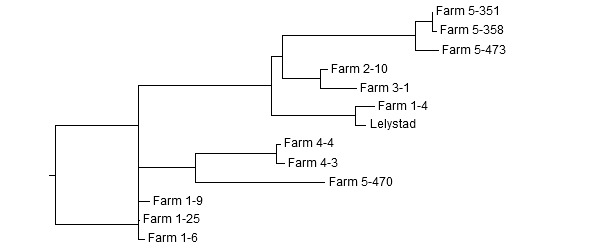
Rycina 2. Dendrogram sekwencji ORF5 uzyskanych z 5 różnych ferm. Przykład interpretacji: w fermie 1 krążą dwa różne szczepy. Trzy sekwencje są podobne do siebie >99%, podczas gdy czwarta sekwencja jest podoba do pozostałych w ~83%. Jest ona za to blisko spokrewniona z sekwencją szczepu Lelystad. Szczepy z ferm 2 i 3 są blisko spokrewnione ze soba (98,2% podobieństwa). Dwa szczepy z fermy 4 są ze sobą blisko spokrewnione (99,5% podobieństwa). W fermie 5 krążą dwa niespokrewnione szczepy. Trzy sekwencje są podobne do siebie w >98%, i jedynie w 81% do czwartej sekwencji.
Tabela: Procentowe podobieństwo sekwecji ORF5 PRRSV Typ 1 z pięciu ferm.
| 1-4 | 1-6 | 1-9 | 1-25 | 2-10 | 3-1 | 4-4 | 4-3 | 5-351 | 5-358 | 5-470 | 5-473 | Lelystad | |
| *** | 83.5 | 83.3 | 83.7 | 93.2 | 92.6 | 86.1 | 86.0 | 88.1 | 88.0 | 83.2 | 88.3 | 98.7 | 1-4 |
| *** | 99.2 | 99.7 | 84.2 | 83.0 | 86.0 | 86.0 | 81.4 | 81.2 | 86.3 | 81.4 | 82.1 | 1-6 | |
| *** | 99.5 | 84.5 | 83.3 | 85.6 | 85.6 | 81.4 | 81.2 | 86.5 | 81.4 | 81.9 | 1-9 | ||
| *** | 84.5 | 83.3 | 86.0 | 86.0 | 81.7 | 81.5 | 86.8 | 81.7 | 82.2 | 1-25 | |||
| *** | 98.2 | 86.6 | 86.5 | 91.1 | 90.9 | 84.2 | 90.4 | 93.3 | 2-10 | ||||
| *** | 84.4 | 84.2 | 90.2 | 90.0 | 83.3 | 89.7 | 92.9 | 3-1 | |||||
| *** | 99.5 | 82.5 | 82.7 | 90.6 | 82.5 | 84.8 | 4-4 | ||||||
| *** | 82.3 | 82.5 | 90.9 | 82.2 | 84.6 | 4-3 | |||||||
| *** | 99.8 | 81.0 | 98.3 | 88.4 | 5-351 | ||||||||
| *** | 81.2 | 98.2 | 88.2 | 5-358 | |||||||||
| *** | 80.9 | 83.2 | 5-470 | ||||||||||
| *** | 88.8 | 5-473 | |||||||||||
| *** | Lelystad |
Interpretacja wyników porównania sekwencji PRRSV
Sekwencja ORF5 PRRSV jest zbudowana z około 600 nukleotydów. Różne badania określają częstotliwość występowania mutacji w tym genie na około 0,5% do 1% rocznie. Zmienna szybkość z jaką dochodzi do mutacji zależy od różnych czynników nie związanych z samym wirusem. Poziom nieswoistej i swoistej odporności na zakażenie PRRSV ma olbrzymi wpływ na replikację wirusa a jej wysoki poziom ogranicza liczbę jego cząstek i w efekcie transmisję między zwierzętami. Niższe siewstwo oznacza niższy poziom transmisji, co w efekcie pogłębia dalsze ograniczenie replikacji wirusa co skutkuje również zmniejszeniem szybkości z jaką dany szczep mutuje. Ten sam szczep wirusa PRRS w różnych warunkach (poziom odporności świń) może ulegać mutacjom z różną prędkością. Tak więc niekiedy obserwujemy częstotliwość występowania mutacji niższą lub wyższą niż podawany zakres 0,5-1% rocznie.
Podstawowym pytaniem w analizie genetycznej PRRSV jest czy dwie sekwencje są blisko ze sobą spokrewnione (należą do dwóch wariantów tego samego szczepu) czy też są niezależne (należą do dwóch niespokrewnionych szczepów). Powszechnie przyjmuje się granicę 97-98% podobieństwa, poniżej której szczepy uznaje się za niespokrewnione ze sobą. Należy pamiętać jednak, że sztywne trzymanie się tej granicy, bez wzięcia pod uwagę dodatkowych elementów, może prowadzić do błędnych wniosków. Różnice między wariantami tego samego szczepu krążącego w populacji przez kilka lat w końcu będą większe niż 2-3%. Aby analiza mogła doprowadzić do użytecznych wniosków należy w badaniach wykorzystać więcej informacji takich jak na przykład daty i geograficzna lokalizacja ferm, z których pochodziły badane wirusy. Jest bardzo ważne aby analizę nowych szczepów PRRSV rozpatrywać na tle zestawu sekwencji reprezentatywnego dla danej fermy, systemu produkcyjnego czy regionu, a także reprezentującego globalny zakres zmienności genetycznej wirusa.
Sekwencjonowanie PRRSV może wykazać bliskie pokrewieństwo lub jego brak między dwoma szczepami (Rycina 2) lecz nie może być wykorzystywane do przewidywania lub wyjaśniania zjawisk odpornościowych w stadach zakażonych PRRSV. Sekwencjonowanie PRRSV nie pozwala również na przewidywanie przebiegu klinicznego zakażenia danym szczepem ponieważ jak dotychczas nie zdefiniowano genetycznych markerów zjadliwości wirusa.
Obecnie prowadzonych jest wiele regionalnych programów zwalczania PRRS lub eliminacji wirusa. Mają one miejsce głównie w Ameryce Północnej ale również w Europie. Pełny obraz zmienności PRRSV na początku realizacji programu jest krytyczny dla skutecznego monitoringu postępów i skuteczności postępowania, i dla identyfikacji pojawienia się nowych szczepów wirusa w fermach lub regionach.